
Subtitle: This time… its correlated.
Previously I showed that with the default ML snp calling on GBS data, heterozygosity was higher with high and low amounts of data. I then took my data, fed it through a snp-recaller which looks for sites that were called as homozygous but had at least 5 reads that matched another possible base at that position (i.e. a base that had been called there in another sample). I pulled all that data together, and put it into a single table with all samples where I filtered by:
<%50 heterozygosity,
>%95 coverage,
>5% minor allele frequency
I plotted heterozygosity in each sample there versus number of reads. In the last post I used the size of the fastq files, but it is comparable.
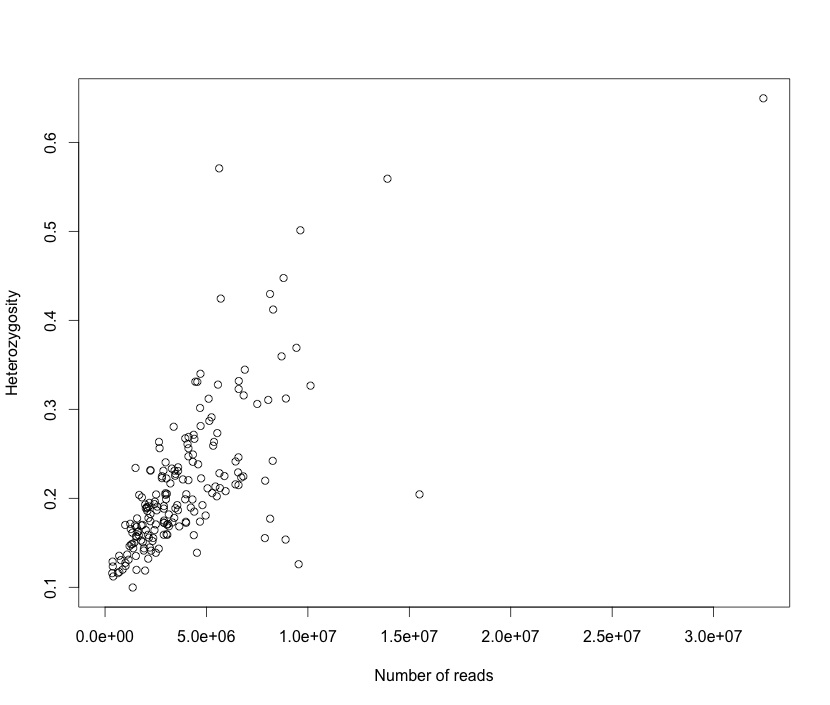
I didn’t put a trend line because you don’t even need to. It is grim. The recalling script is not conservative enough when calling heterozygotes. It’s possible that the aligner isn’t working well enough and so misalignments are giving it the fodder to call heterozygotes on everyone. The main thing is that it is clear that we can’t just call every site with one read supporting an alternate allele as a het and then filter by overall heterozygosity.