Appendix 2
DNA-Binding Assays
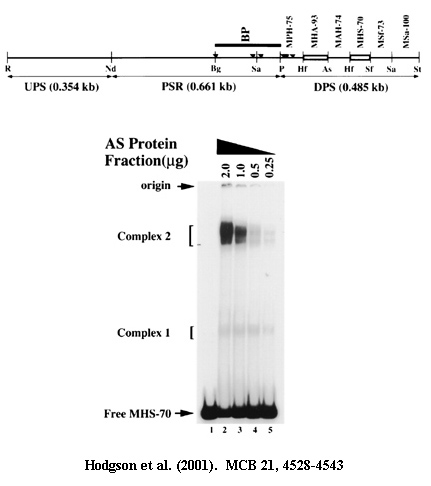
Electrophoretic mobility shift assays (EMSAs): This is the assay you will see more often than any other, and one of the techniques you have to know backwards and forwards. A DNA of a given size has a characteristic electrophoretic migration pattern on a given gel. If that DNA can be bound by a protein, then the protein-DNA mixture will migrate more slowly than the control DNA that has not been incubated with protein. This change in relative mobility is called a mobility shift. Indeed, this assay is often referred to as a gel retardation experiment. Normally the DNA is an end-labelled restriction fragment, present at very low concentration, and the protein is present in excess.
EMSAs have many uses. If, for example, you wish to determine where proteins bind a regulatory region, you can simply make fragments that span the region of interest, test each individually and determine which fragments show mobility shifts. If you wish to determine if a given transcription factor (TF) binds a given DNA fragment, you incubate the two together and look for the presence of a mobility shift on a gel. If you wish to determine which domain in a protein is responsible for DNA recognition, make mutations in the protein, express them and test the mutant proteins for their ability to mobility shift their targets.
Supershift assays: A mobility shift assumes that a protein binds a DNA fragment. If an additional protein binds the first protein, or can also bind the same DNA fragment, then the mobility of this three part complex will be slower than the mobility of the DNA and the first protein, and naturally the mobility of this will be slower than that of the DNA fragment alone. Therefore, this extra retardation is called a supershift.
Antibody supershift assays: Suppose you have antibodies to a variety of TFs and you want to determine if one of the TFs binds a certain DNA target. You can take a nuclear extract, incubate it with a DNA fragment and show that there is a mobility shift. Then you repeat the experiment, adding a different antibody each time. If you see a supershift, you know you have identified the TF responsible for the initial mobility shift. Another example occurs when two different TFs can bind to the same target. You might wish to know if they can both bind the target at the same time, or whether they are in competition, so that one can bind but not the other. In this latter case, you would do EMSAs with each protein individually and compare the result to an EMSA done with a mixture of the two proteins. If you see a supershift, you can be certain that both TFs bind the same target simultaneously.
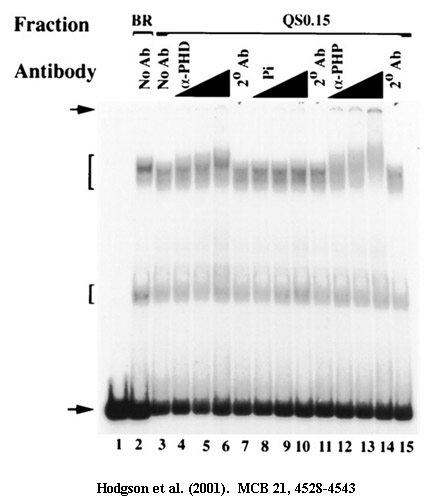
Mobility shift (for DNA-binding proteins): This is a variation on a supershift. If two proteins, A and B bind the same DNA target as homodimers (ie. A-A and B-B), and they are of different molecular weights, then two different bands will be seen after EMSA if they are added separately to the target. If the proteins are now mixed and added to the target, there will be only the same two bands if the proteins do not interact, and cannot both bind the DNA at the same time. If the two proteins bind independently to the target, there will be a new mobility shift that is of higher molecular weight than either single protein. However, if A and B can form heterodimers (A-B), there will be a new band of intermediate mobility.
Competition assays: Thus far we have discussed experiments in which pure proteins or extracts are added to labelled DNA fragments. However, much can be learned from adding unlabelled competitor DNA in increasing amounts to the reaction. Below are several uses of competition assays.
1. It should be obvious that if the competitor is exactly the same as the target, the proportion of protein bound to the labelled DNA compared to the proportion bound to the unlabelled DNA will be equivalent to the ratio of labelled to unlabelled DNA. This if enough unlabelled (or cold) competitor is added, binding to the labelled (or hot) target will be undetectable. So, if binding to a given target is specific, two predictions follow: i) it should be possible to compete away binding to the target by adding cold target DNA; and ii) it should NOT be possible to compete away activity by adding non-specific unlabelled DNA. If it is possible to compete with random DNA, then this suggests that binding of the factor to the DNA is non-specific.

2. It is possible to make mutations in the unlabelled competitor DNA and compare the efficiency of competition with the mutant and wildtype DNA. If the mutation decreases the affinity of the factor for the DNA, then it will be necessary to add more unlabelled mutant competitor relative to wildtype to achieve abolition of binding to the labelled target. Moreover, the ratio of the amount of mutant to wildtype can provide a quantitative estimate of relative change in binding affinity. If you have to add ten times as much mutant competitor to abolish binding to a labelled target relative to the wildtype competitor, then you can surmise that binding to the mutant is 10X less than the binding to the wildtype. Conversely, binding to the wildtype fragment is 10X more than to the mutant. The same idea can be used to compute the relative affinity of a TF for two different natural binding sites.

DNA Footprinting: This is another DNA-binding assay but it gives precise information about where a protein binds DNA. Normally EMSA is used to narrow down the region followed by DNA footprinting. The target DNA is end-labelled and is then incubated with a purified protein or protein extract. Afterwards, an endonuclease or chemical cleavage agent is allowed to cut the DNA at low concentration, so that on average, only one cut is made per DNA molecule. The idea is that every base pair can be attacked except those that are protected by the bound protein(s). The protein is removed and the labelled DNA is fractionated on a sequencing gel. There will be a ladder corresponding to the DNA sequence and there will be a hole in the ladder where the protein was bound. This is a DNA footprint. Comparison of the footprint to a normal sequencing reaction allows you to identify, more or less to the base pair, where a protein binds.


Cloning a TF if the binding site is known: It often happens that during EMSA and DNA footprinting studies of a regulatory region, a given sequence is known to bind a regulatory protein but the identity of the molecule is unknown. How can the TF that binds the sequence by identified? There are two methods and both depend on affinity. One method is to make a column containing many copies of the target sequence and to use this as an affinity column to purify the protein(s) that bind to it by chromatographing a nuclear extract. The alternative is to screen a cDNA expression library, using labelled target DNA as a probe. The idea is that only a colony containing a cDNA that encodes for the binding protein will be able to bind the target DNA, and this can be identified using autoradiography. Once the colony is purified, you will have the cDNA for the TF of interest.
Determining DNA-binding sequence of a TF: The converse of above also happens. Sometimes a protein with a putative DNA-binding domain is identified but its normal target is unknown. How can the recognition sequence be determined? The problem is that the genome is large and that many similar but slightly different sequences might be recognized by the TF. An ingenuous solution has been found by combining the ability to synthesize random oligonucleotides in large quantities and PCR. If oligos are synthesized at random, a quick calculation will show you that the concentration of any one oligo becomes very small the longer the oligo gets {P=(1/4)n}. The method is to make an affinity column with the TF, and then chromatograph the random oligos. The majority will wash through the column and some will be retained. These are eluted from the column, amplified by PCR and then rechromatographed. Each round of chromatography enriches for high affinity DNA-binding sites. These enriched fragments can be sequenced and the consensus binding site can be determined. With this sequence in hand, it is then possible to use this sequence as a probe to look for genes with the corresponding regulatory regions (these days this is accomplished through DNA microarrays).