
Evolution in Finite Populations
Up until now, we have treated populations as being infinitely large.
For example, we have used equations to determine p[t+1] assuming that each type produces EXACTLY as many offspring as predicted by their fitnesses.
In any finite population, however, individuals may have more or fewer offspring than expected, simply by chance.
The chance increases or decreases in the frequency of an allele in a finite population are called random genetic drift.
How does random genetic drift affect evolution in the absence of selection?
How does random genetic drift affect evolution in the presence of selection?
Example: A Birth-Death Model.
Imagine a population of 1000 haploid individuals, half of which are A and half a.
At any point in time, any individual may die. It is then replaced by the reproduction of another individual chosen at random from the population.
At first, when the allele frequency remains near p=1/2:
 No change in number of A alleles.
No change in number of A alleles.
Number of A alleles decreases by one.
Number of A alleles increases by one.
No change in number of A alleles.
In this example, A and a are equally fit; they have an equal chance of dying and an equal chance of reproducing.
The class has simulated this model.
Of the birth-death events leading to a change in the number of A individuals, 50% (heads) led to an increase by one and 50% (tails) led to a decrease by one.
Consequently, the frequency of A varied over time from 1/2 by random drift.
(What would soon go wrong with our "simulation"?)
A more commonly used model of random genetic drift is called the Wright-Fisher Model, which assumes:
Imagine labelling each allele in a population at some point in time. These "alleles" will drift up and down in frequency, until eventually only one remains.
Any one of these alleles has an equal chance of being the "lucky" allele that fixes.
In a haploid population of size 5, what is the chance that allele #1 fixes?
In a haploid population of size N, what is the chance that any particular allele copy fixes?
If there are n copies of allele A and N-n copies of allele a in a haploid population, what is the probability that allele A eventually fixes?

In the absence of mutation and selection, allele frequencies drift up and down in frequency until, eventually, one allele becomes fixed.
We have been discussing the Wright-Fisher process for a haploid population, but the same method works for a diploid population as long as alleles within an individual are randomly sampled from a gamete pool.
In a diploid population with N individuals, there will be 2N alleles.
What is the chance that any particular allele copy fixes?
If there are n copies of allele A and 2N-n copies of allele a in a diploid population, what is the probability that allele A eventually fixes?
In the example shown above, it took nine generations for the third allele to fix.
In general, the AVERAGE amount of time it will take for a single allele to fix within a population is twice the number of alleles within a population:
2N generations with N haploid individuals
4N generations with N diploid individuals
ASIDE: The time to fixation of an allele A is approximately the same whenever A is initially rare (small p[0]).
For example, in your PopBio assignment, p=0.1 and N=10 (diploid population), what fraction of the time should A fix? How long should it take for A to fix when it does fix?

If it takes, on average, 4N generations for a single allele to spread to fixation within a diploid population (forwards in time), how long ago in the past, on average, must we look before all the alleles currently present in the population shared a common ancestor?
[This is called the coalescence time.]
We can look at this process forward or backwards in time!
Random genetic drift causes, on average, a loss of genetic variation within a population.
This is obviously true in the long-term, since a population eventually drifts to fixation on one allele.
For example, Buri (1956) examined 107 Drosophila populations that each started with 16 heterozygotes for a brown eye mutation (bw):

The loss of variability can be measured by changes in the "Expected Homozygosity" (f) of a population.
Expected Homozygosity (f) = The probability that two alleles drawn at random are the same allele.
For instance, with two alleles at a locus, the expected homozygosity will initially equal:
since p2[0] is the probability that both the first and the second allele chosen at random are A and q2[0] is the probability that both the first and the second allele are a.
In generation t, when two offspring alleles are chosen from the population at random, there are two possibilities:

(1) What is the probability that both offspring alleles come from the same parental allele in a diploid population with 2N alleles?
(2) What is the probability that both offspring alleles come from different parental allele in a diploid population with 2N alleles?
In the first case, homozygosity becomes one (the two alleles ARE same allele in the previous generation).
In the second case, the two offspring alleles will be the same allele if they are descended from parents with the same allele, which is given by the expected homozygosity in the previous generation, f[t-1].
The smaller the population, the more likely that two alleles will be the same just because they had the same parent.
The expected homozygosity rises in all finite populations, but rises fastest in small populations.
As expected homozygosity rises, the amount of genetic variation within the population declines. The amount of genetic variation is measured by:
Expected Heterozygosity (H) = The probability that two alleles drawn at random are different alleles: H = 1 - f.
In generation t,
= 1 - 1/(2N) - (1-1/(2N)) f[t-1]
= (1-1/(2N)) H[t-1]
The expected heterozygosity within a diploid population declines at a rate 1/(2N) each generation.

REMEMBER: These are only averages. In any particular population, heterozygosity will rise and fall over time, eventually reaching zero.

Even in small populations, heterozygosity does not disappear forever, since mutations continually arise.
Two alleles drawn at random are NOT the same if either mutates to a new allele.
Therefore, for two alleles drawn at random to be the same, they must both be non-mutant:
 )2
)2
where is the mutation rate to new neutral alleles.
At equilibrium, the loss of genetic variability by drift and the gain by mutation counter-balance, so that on average:
+ 1)
H* = 4 N /(4 N + 1)
[CHALLENGE: Derive these]
These are only averages. Any particular locus may or may not be fixed.
Not only do new mutations contribute to the amount of heterozygosity within a population, but occasionally these new mutations rise to fix within a population.
This creates a constant turn-over in the alleles carried by a population, even in the absence of selection.
In a diploid population, how many new mutations appear each generation?
For any one of these mutations, what is the probability that it will be the "lucky" allele from which the entire population will eventually descend?
The turn-over of neutral alleles will occur at a rate equal to 2N/(2N) = !
This result does not depend on the population size.
 |
Motoo Kimura derived this result and used it to predict that the number of DNA substitutions that occur within the genome should rise as a linear function of time. |
Comparing  -globin genes from various vertebrates, Kimura showed (1983) that this prediction matched the inferred numbers of amino acid substitutions:
-globin genes from various vertebrates, Kimura showed (1983) that this prediction matched the inferred numbers of amino acid substitutions:
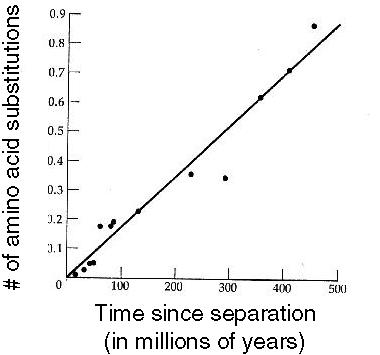
The number of substitutions can be used as a MOLECULAR CLOCK, indicating how much time has passed.
Would we expect a similar rate of evolution if selection were also acting?
Haldane (1927) showed that, in a large population, the probability of a new mutant allele fixing is 2s, where s is its selective advantage.
Kimura noted that the rate of fixation of new adaptive mutants that arise at rate would therefore equal 2N*(2s) in a diploid population.
This leads to a prediction: A gene sequence should evolve at a constant rate over a range of population sizes ONLY if very little selection is acting on that sequence.
When would you expect the fate of an allele to depend more on random genetic drift than on selection?
When would you expect the fate of an allele to depend more on selection than on random genetic drift?
4Ns determines the relative roles of selection (important when 4Ns>>1) and random genetic drift (important when 4Ns<<1).
SOURCES:
 Back to Biology 336 home page.
Back to Biology 336 home page.